En la última década, la caducidad de las patentes que protegen los anticuerpos monoclonales biosimilares terapéuticos abrió una oportunidad para el desarrollo y aprobación de versiones biosimilares de estos fármacos. La complejidad de estas moléculas biológicas requirió la imposición de regulaciones estrictas para establecer una robusta comparabilidad con el anticuerpo de referencia en datos fisicoquímicos, analíticos, biológicos y, cuando se consideró necesario, clínicos.
1. Introducción
1.1 La evolución de los anticuerpos monoclonales frente a medicamentos biológicos
Los anticuerpos, también conocidos como inmunoglobulinas, son glicoproteínas complejas producidas por las células B contra sustancias extrañas como parte de la respuesta inmunitaria adaptativa [ 1 , 2 ]. La invención de la tecnología de hibridomas en 1975 por Köhler y Milstein permitió la producción de anticuerpos monoclonales con una especificidad deseada a partir de un clon único de células B [ 3 ].]. A diferencia de los anticuerpos policlonales, los anticuerpos monoclonales son homogéneos, monoespecíficos y pueden producirse en cantidades ilimitadas en el laboratorio. Dado que pueden dirigirse contra casi cualquier epítopo molecular, los anticuerpos monoclonales se adoptaron pronto como una herramienta de diagnóstico, pero tomó más de una década hasta la aprobación de Muromonab-CD3 (Orthoclone Okt3®), que es el primer anticuerpo monoclonal desarrollado con el hibridoma. tecnología comercializada para uso terapéutico [ 4 ]. Sin embargo, dado que los anticuerpos de la tecnología de hibridoma solo tienen secuencias murinas, en pacientes humanos exhibieron una función efectora limitada [ 5 ], fueron anticuerpos anti-ratón inductores inmunogénicos y tuvieron una vida media significativamente reducida [ 6 ].]. Por lo tanto, no fue hasta el desarrollo de anticuerpos monoclonales recombinantes en las décadas de 1980 y 1990 que comenzó una nueva era de la terapia biológica, con los anticuerpos quiméricos [ 7 ], humanizados [ 8 ] y completamente humanos [ 9 ].]. Cada paso implicó el reemplazo gradual de segmentos murinos de la secuencia del anticuerpo por la secuencia humana correspondiente: en los anticuerpos quiméricos se reemplazó la región constante, y en los anticuerpos humanizados, se reemplazó el marco que flanqueaba las regiones determinantes de la complementariedad y la región constante, y en anticuerpos monoclonales humanos la secuencia completa es humana. La ingeniería adicional permitió su personalización, creando variantes en valencia, tamaño, funciones efectoras y con la conjugación de compuestos para su administración a tipos de células objetivo, como el cáncer.
1.2 El surgimiento de los anticuerpos biosimilares y América Latina
En los últimos veinte años, los anticuerpos monoclonales terapéuticos se han aprobado cada vez más y de manera constante y, para 2021, se estima que se habrán aprobado 106 anticuerpos monoclonales en los Estados Unidos o la Unión Europea para el tratamiento de un espectro cada vez mayor de enfermedades [ 10 ]. La aparición de anticuerpos monoclonales terapéuticos de próxima generación en la última década coincide con la expiración de las patentes que protegen los primeros anticuerpos monoclonales recombinantes [ 11 ]. La aprobación en 2013 del biosimilar de infliximab Remsima® [ 12] abrió un campo emergente de competencia en todo el mundo, con el desarrollo de copias biológicas que exhiben una calidad y eficacia equivalentes en comparación con los anticuerpos originales. También fue una oportunidad para que las empresas biofarmacéuticas de América Latina ingresaran a este mercado, alentadas también por sus gobiernos. Sin embargo, las modificaciones postraduccionales de los anticuerpos monoclonales incluyen diferentes grados de glicosilación, variantes de puentes disulfuro o modificaciones del terminal C/N que dependen del proceso de fabricación [ 13 ].]. Debido a esta complejidad estructural, las agencias reguladoras en América Latina pasaron por cambios profundos en sus estándares con el fin de actualizar los criterios de evaluación y aprobación de biosimilares de anticuerpos, lo que requiere análisis de comparabilidad en seguridad y eficacia. Hoy en día, sus requisitos suelen incluir el suministro de información fisicoquímica, farmacéutica y biológica detallada sobre los atributos críticos de calidad del principio activo y el proceso de fabricación. Además, la comparabilidad también requiere establecer si existen variaciones en el tipo de célula huésped a producir de la proteína recombinante, la secuencia de aminoácidos, la estructura secundaria, terciaria y cuaternaria, las interacciones, las modificaciones postraduccionales, la formulación, así como así como impurezas relacionadas con el proceso o almacenamiento. El desafío para su aprobación por parte de las agencias reguladoras es remodelar la accesibilidad de estos costosos medicamentos en América Latina. Aquí enfocamos nuestro análisis en biosimilares que han sido caracterizados en sus propiedades fisicoquímicas y mostraron evidencia de calidad, eficacia y seguridad publicada en la literatura científica, y no incluiremos productos conocidos como copias o copias previstas cuyos patrocinadores no hayan presentado evidencia suficiente de su equivalencia con el producto de referencia.
1.3 Regulación de biosimilares en América Latina
Con el objetivo de cumplir con los estándares internacionales para la producción y desarrollo de medicamentos biológicos, desde 2008, los países de América Latina comenzaron a unirse al Esquema de Cooperación de Inspección Farmacéutica (PIC/S) y hoy Argentina, Brasil y México son miembros. Esta organización establece estándares en el desarrollo internacional, implementación y mantenimiento de Buenas Prácticas de Manufactura (GMP) armonizadas y sistemas de calidad de inspecciones de medicamentos. Solo estos tres países de América Latina han desarrollado una industria biotecnológica que incluye empresas privadas con capacidad para fabricar medicamentos biológicos. Mientras tanto, hoy en día la mayoría de los países de América Latina han aprobado regulaciones específicas para el registro de medicamentos biológicos y de productos bioterapéuticos similares o biosimilares.14 ]. Como era de esperar, cada país de América Latina ha adoptado su propio marco regulatorio para el registro y aprobación de biosimilares.
El registro de medicamentos biosimilares en Argentina está controlado por la Administración Nacional de Medicamentos, Alimentos y Dispositivos Médicos (Administración Nacional de Medicamentos Alimentos y Tecnología Médica, ANMAT). En 2011 se publicó la disposición N° 7729/2011, que “aprobó los requisitos y lineamientos para el registro de especialidades medicinales de origen biológico cuya composición cualitativa-cuantitativa, indicación terapéutica y vía de administración propuesta, tengan precedentes en otras especialidades medicinales de origen biológico autorizados y registrados ante esta Administración u otra Autoridad Sanitaria Reguladora (medicamento biológico de referencia o comparador), del cual se tiene evidencia de comercialización efectiva y caracterización suficiente de su perfil riesgo-beneficio” [ 15]. El término “biosimilar” no se utiliza en ninguna de las disposiciones reglamentarias de ANMAT aprobadas a la fecha [ 16 ], refiriéndose a estos productos como “medicamentos biológicos similares”.
En Brasil, la Agencia Nacional de Vigilancia Sanitaria (Agencia Nacional de Vigilancia Sanitaria, ANVISA) es la agencia de registro a cargo de la aprobación de los biosimilares, que está regulada por la resolución RDC 55/2010 [ 17 , 18 ]. Si bien ANVISA no utiliza el término “biosimilar” en su resolución, su definición se fusiona con el término “producto biológico”, que se define como el medicamento biológico no nuevo o conocido que contiene una molécula con actividad biológica conocida, ya registrada en Brasil y que ha pasado por todas las etapas de fabricación (formulación, llenado, liofilización, etiquetado, envasado, almacenamiento, control de calidad y liberación del lote de producto biológico para su uso) [ 17]. La aprobación de estos productos biológicos requerirá estudios de comparabilidad con un comparador biológico en cuanto a parámetros clínicos y no clínicos basados en la calidad, eficacia y seguridad, con el fin de establecer que no existen diferencias detectables en términos de calidad, eficacia y seguridad entre los productos. El medicamento biológico de referencia o innovador recibe el nombre de “nuevo medicamento biológico”, y el producto de referencia “producto biológico comparador” es un producto biológico que ya ha sido registrado en la ANVISA sobre la base de un expediente completo y ya ha sido comercializado en el país.
La Agencia Nacional de Medicamentos (ANAMED) es el organismo regulador en Chile que regula la norma técnica para el registro sanitario de productos biotecnológicos derivados de técnicas de ADN recombinante. En su norma técnica reguladora de medicamentos biológicos, el término biosimilar se define como “el medicamento biotecnológico que ha demostrado ser comparable en calidad, seguridad y eficacia al producto biotecnológico de referencia, con base en su caracterización exhaustiva mediante estudios de comparabilidad en igualdad de condiciones, consistente en estudios de calidad y estudios no clínicos y clínicos, todos ellos comparativos” [ 19 , 20 ].
El organismo regulador responsable de la aprobación de medicamentos biológicos en Colombia es el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA). En 2014 se publicó el Decreto N° 1782 [ 21 ] que describe la vía de registro de biosimilares en ese país. Si bien la directiva no utiliza el término “biosimilar”, se refiere a ellos como “productos bioterapéuticos similares” y establece un régimen normativo específico para su registro. Esta aplicación requiere una serie de pruebas que comparen los atributos de calidad, seguridad y eficacia entre el biosimilar y el medicamento biológico de referencia para demostrar que el medicamento en evaluación es muy similar al medicamento de referencia [ 21 ].
La regulación de los medicamentos biológicos en el Ecuador es supervisada por la Agencia Nacional de Regulación, Control y Vigilancia Sanitaria (ARCSA). El Ministerio de Salud aprobó en 2019 el acuerdo 385 que regula la comercialización de medicamentos biológicos para uso y consumo humano en el Ecuador, así como para establecer el procedimiento general para la obtención del Registro Sanitario. En esta directiva, los biosimilares se definen como un medicamento biológico que, mediante el ejercicio de comparabilidad, ha demostrado ser similar en términos de calidad, seguridad y eficacia al medicamento biológico de referencia [ 22 , 23 ].
México es otro país donde el término “biosimilar” no se utiliza en sus normas regulatorias para la aprobación de medicamentos biológicos. La Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS) es la agencia en México responsable de regular la aprobación, fabricación y comercialización de medicamentos biológicos. La norma NOM-257-SSA1-2014 establece el marco regulatorio de los medicamentos biotecnológicos y se refiere al “medicamento biotecnológico biocomparable”, como el medicamento biotecnológico no innovador que demuestra ser comparable en términos de seguridad, calidad y eficacia del medicamento biotecnológico de referencia. a través de estudios de biocomparabilidad [ 24 , 25 ].
El registro de medicamentos biológicos en Paraguay está regulado por la Dirección Nacional de Vigilancia Sanitaria (Dirección Nacional de Vigilancia Sanitaria, DINAVISA). El Decreto N° 6611 aprobado en 2016 estableció los requisitos para la aprobación de medicamentos biológicos e incluye la definición de medicamentos biológicos similares o biosimilares [ 26 ]. En este decreto, los biosimilares se definen como un medicamento biológico que demuestra similitud en términos de seguridad, calidad, eficacia e inmunogenicidad con el medicamento biológico de referencia a través del ejercicio de comparabilidad [ 26 ].
En Perú, la Dirección General de Medicamentos, Insumos y Drogas (DIGEMID) es el organismo encargado de la regulación y normativa en materia de aprobación y certificación de medicamentos biológicos. En 2016 se aprobó el Decreto Supremo N° 013-2016-SA que regula el registro de productos biológicos, que opten por la vía de la similitud, o productos biológicos similares [ 27 ]. En esta norma, se definen como el producto biológico, que en términos de calidad, seguridad y eficacia, es similar a un producto biológico de referencia [ 27 ].
La mayoría de los países restantes de América Latina no cuentan con agencias dedicadas o normas específicas que regulen la aprobación y vigilancia de biosimilares, por lo que no se incluirán en este análisis [ 25 ].
2. Anticuerpos monoclonales biosimilares aprobados en América Latina
Actualmente existen cinco anticuerpos monoclonales terapéuticos registrados en América Latina cuyas patentes expiraron en los últimos años y cuentan con versiones biosimilares comercializadas en la región ( Figura 1 ). Se trata de rituximab, trastuzumab, infliximab, adalimumab y bevacizumab, y solo Argentina, Brasil y Colombia cuentan con al menos una versión biosimilar aprobada para cada anticuerpo monoclonal ( Figura 1 ). Con más de diez biosimilares aprobados, Argentina y Brasil son los países de América Latina con más anticuerpos monoclonales biosimilares aprobados. Le siguen Colombia, Perú, Paraguay, México y Chile, con 3 a 5 biosimilares de anticuerpos monoclonales, y la menor adopción de biosimilares se encuentra en Ecuador, Bolivia y Uruguay ( Cuadro 1 y Figura 1 ).). Informes recientes indican que en Brasil los precios en dólares estadounidenses de los productos biológicos originales, incluidos los anticuerpos monoclonales terapéuticos, han disminuido significativamente en la última década. La aparición de la competencia de los biosimilares, con sus precios más bajos, puede fortalecer esta tendencia [ 28 ]. Se espera que la aprobación de más anticuerpos monoclonales biosimilares aumente la competencia, disminuya los costos de atención médica y amplíe la accesibilidad de esta clase de medicamentos.
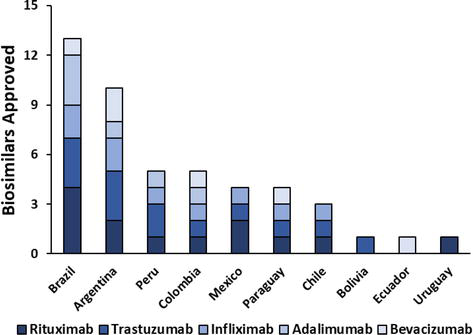
Figura 1.
Gráfica comparativa del número de biosimilares aprobados en países de América Latina.
| Nombre común | Nombre del anticuerpo | Marca (país) | Fabricante (país) | Distribuidor (país) |
|---|---|---|---|---|
| Rituximab | # Rituxan® / # Mabthera® | Roche (SO) | Roche | |
| PF-05280586 | Ruxience® (BR) | Pfizer (Estados Unidos) | Wyeth (BR) | |
| RTXM83 | Novex® (AR, PA, UR) Rigetuxer® (ME) | mAbxencia (AR) | Laboratorios Elea (AR) Laboratorios PISA (ME) Laboratorios Bioéticos (PA) Urufarma (UR) | |
| Vivaxxia® (BR) | Libbs (BR) | Libbs (BR) | ||
| GP2013 | Rixathon® (AR) Riximyo® (BR) Arasamila® (ME) | Sandoz (AU) | Novartis (AR) Sandoz (BR, ME) | |
| CT-P10 | Truxima® (BR, CH, CO) | Celltrion (SK) | Celltrion (BR, CO) Saval (CH) | |
| Zytux | Zaytux® (PE) | AryoGen (IR) | PeruLab (PE) | |
| Trastuzumab | # Herceptin® | Roche (SO) | Roche | |
| ABP 980 | Kanjinti® (AR, PE) | Amgen (Estados Unidos) | Varifarma (AR, PE) | |
| MYL-1401O | Ogivri® (CO) Tuzepta® (AR) Zedora® (BR) Bisintex® (BO, CH, PA, PE) | Biocon (IN) | Laboratorios Raffo (AR) Libbs (BR) PharmaTech Boliviana (BO) Recalcine (CH) Mylan (CO) Pharma International (PA) Abbott (PE) | |
| PF-05280014 | Trazimera® (AR, BR) | Pfizer (Estados Unidos) | Pfizer (AR) Wyeth (BR) | |
| CT-P6 | Herzuma®(BR) | Celltrion (SK) | Celltrion (BR) | |
| infliximab | # Remicade® | Janssen (Estados Unidos) | Janssen | |
| CT-P13 | Remsima® (AR, BR, CH, CO, EC, ME, PA) Flixceli® (PE) | Celltrion (SK) | Gobbi-Novag (AR) Celltrion (BR, CO, ME, PA) Saval (CH) Oxialfarm (EC) AC Pharma (PE) | |
| PF-06438179 / GP1111 | Ixifi® (AR) Xilfya® (BR) | Pfizer (Estados Unidos) | Pfizer (AR) Wyeth (BR) | |
| Adalimumab | # Humira® / # Trudexa® | AbbVie (EE. UU.) | AbbVie | |
| ABP 501 | Amgevita® (AR, BR, CO, PE) | Amgen (Estados Unidos) | Amgen (AR, BR, CO) TecnoFarma (PE) | |
| GP2017 | Hyrimoz® (BR) | Sandoz (GE) | Sandoz (BR) | |
| PF-06410293 | Xilbrilada® (BR) | Pfizer (Estados Unidos) | Wyeth (BR) | |
| Bevacizumab | # Avastin® | Roche (SO) | Roche | |
| BEVZ92 | Bevax® (AR, EC, PA) | mAbxencia (AR) | Laboratorios Elea (AR) Grünenthal (EC) Laboratorios Bioéticos (PA) | |
| ABP 215 | Mvasi® (AR, BR, CO) | Amgen (Estados Unidos) | Amgen (CO) |
Tabla 1.
Anticuerpos monoclonales biosimilares aprobados en América Latina.
Anticuerpos monoclonales de referencia. Países: Argentina (AR); Austria (AU); Bolivia (BO); Brasil (BR); Chile (CH); Colombia (CO); Ecuador (CE); Alemania (GE); India (IN); Irán (RI); México (ME); Paraguay (PA); Perú (PE); Rusia (RU); Corea del Sur (SK); Suiza (SO); Uruguay (RU); y Estados Unidos (EE.UU.).
2.1 Rituximab
Desarrollado por Genentech en los Estados Unidos, el rituximab se comercializa con el nombre de marca Rituxan® (también conocido como MabThera®) y actualmente lo comercializa Roche. Rituximab es un anticuerpo monoclonal quimérico murino/humano con isotipo IgG1/κ dirigido contra el antígeno CD20 expresado por las células B utilizado para el tratamiento del linfoma no Hodgkin (LNH), leucemia linfocítica crónica (LLC) [ 29 ] y reumatoide. artritis [ 30]. Rituximab fue aprobado por la FDA en 1997 para el tratamiento de linfomas de células B y fue el primer anticuerpo monoclonal recombinante quimérico aprobado contra el cáncer. Se han desarrollado varios biosimilares de rituximab a lo largo de los años, y para 2021 hay cinco biosimilares diferentes de rituximab aprobados en América Latina, con nueve marcas diferentes comercializadas en Argentina, Brasil, Chile, Colombia, Ecuador, México, Paraguay, Perú y Uruguay. ( Tabla 1 ).
2.1.1 Ruxience® (Pfizer)
PF-05280586 (Ruxience®) es un biosimilar de rituximab desarrollado en Estados Unidos por Pfizer y comercializado en Brasil como Ruxience® por Wyeth Industria Farmaceutica. Es un anticuerpo monoclonal utilizado en el tratamiento de varios tipos de cáncer e indicaciones inmunológicas. En Brasil, se aprobó el PF-05280586 con las mismas indicaciones terapéuticas aprobadas para el rituximab de referencia.
Se llevó a cabo una caracterización bioquímica y funcional comparativa para determinar el nivel de similitud fisicoquímica, se generaron mapas de péptidos trípticos tanto para PF-05280586 como para rituximab-EU y se resolvieron mediante cromatografía líquida de alta resolución en fase inversa Ryan [ 31 ]. Este estudio demostró que PF-05280586 tiene una secuencia de aminoácidos primaria idéntica a la de rituximab. Además, se demostró que es muy similar en función de la comparación de atributos críticos fisicoquímicos y no clínicos.in vitrocaracterísticas funcionales [ 31 ].
En un estudio aleatorizado de similitud farmacocinética (PK) de 3 vías en sujetos con artritis reumatoide activa, se demostró la equivalencia PK entre PF-05280586 y rituximab-EU, PF-05280586 y rituximab-US, y rituximab-EU y rituximab-US. Este estudio también demostró perfiles comparables de agotamiento de células B positivas para CD19, respuestas farmacodinámicas (PD), seguridad e inmunogenicidad para todos los tratamientos [ 32 , 33 , 34 ].
Se llevó a cabo un estudio de fase III para comparar la seguridad y la eficacia de PF-05280586 versus rituximab-EU en pacientes con linfoma folicular de baja carga tumoral CD20-positivo [ 35 ]. Este estudio demostró que la eficacia de PF-05280586, medida por la tasa de respuesta global, es similar a la de rituximab-EU [ 35 ].
2.1.2 Novex® / Rigetuxer® / Vivaxxia® (mAbxience)
RTXM83 (Novex® / Rigetuxer® / Vivaxxia®) es un biosimilar de rituximab desarrollado en Argentina por PharmADN (hoy mAbxience) y es el primer anticuerpo monoclonal terapéutico biosimilar desarrollado por una empresa biofarmacéutica local en Argentina. RTXM83 se comercializa en Argentina con la marca Novex® por Laboratorios Elea. En Paraguay, RTXM83 se comercializa como Novex® por Laboratorios Bioéticos, y en Uruguay también se vende como Novex® por Urufarma. En México, RTXM83 se comercializa con la marca Rigetuxer ® por Laboratorios PISA. En Brasil, RTXM83 es fabricado por Libbs y comercializado por el mismo laboratorio que Vivaxxia® [ 36 , 37 ].
RTXM83 está autorizado para LNH con ensayo clínico, y por extrapolación para las siguientes indicaciones terapéuticas a LLC, artritis reumatoide, pacientes adultos con granulomatosis de Wegener (GW) y poliangelitis microscópica (PSM).
Los estudios de comparabilidad han mostrado propiedades fisicoquímicas similares entre RTXM83 y el rituximab de referencia en secuencia primaria y enlaces disulfuro, modificaciones de aminoácidos N-terminal y C-terminal, estabilidad térmica, variantes de carga, patrón de glicosilación, presencia de agregados de orden superior, pureza y afinidad de unión al receptor neonatal y otros receptores Fc [ 38 ]. Más estudios de comparabilidad de la actividad biológicain vitrose realizaron, mostrando similitud en las pruebas de potencia de citotoxicidad mediada por células dependientes de anticuerpos (ADCC) y unión al objetivo molecular CD20 [ 39 ]. Además, los estudios in vivo en monos cynomolgus mostraron similitud en la farmacocinética (PK), incluido el área bajo la curva de concentración-tiempo (AUC), la concentración máxima del fármaco y la farmacodinámica (PD), incluida la reducción de las células CD20 y CD40 [ 40 ].
Los datos del ensayo clínico de fase III NCT02268045 en pacientes con linfoma difuso de células B grandes han mostrado similitud al comparar los parámetros farmacocinéticos en pacientes tratados con RTXM83 y con rituximab de referencia (en ambos casos coadministrado con ciclofosfamida, doxorrubicina, vincristina y prednisona – CHOP) [ 41 ]. Además, la EP se evaluó en términos de agotamiento del recuento de células B CD20 positivas y CD19 positivas, duración de la supresión y tiempo de recuperación, con un perfil similar observado para ambos brazos de tratamiento [ 41]. Además, el estudio de fase III, aleatorizado, doble ciego, que comparó RTXM83 frente a rituximab de referencia, ambos en combinación con CHOP, no mostró diferencias evidentes en el perfil de seguridad en términos de naturaleza, frecuencia y gravedad de los eventos adversos, ni en la eficacia en términos de respuesta tumoral. La inmunogenicidad se evaluó como la incidencia de anticuerpos antidrogas, que fue baja y similar entre RTXM83 y el rituximab de referencia, con ≤ 4 % en ambos brazos [ 41 ].
ANMAT en Argentina ha establecido un Registro de Tratamiento prospectivo como parte de su programa de farmacovigilancia para la detección, evaluación, comprensión y prevención de efectos adversos derivados del uso de medicamentos, y en 2014 comenzó a recopilar datos de pacientes tratados con RTXM83. Los médicos enviaron información a este registro entre 2014 y 2017 de pacientes tratados con RTXM83 para LNH folicular, LNH difuso de células B grandes, LLC e indicaciones clínicas no aprobadas [ 42 ]. Este programa de farmacovigilancia activa de RTXM83 permite la monitorización continua del perfil de seguridad de este biosimilar, y su frecuencia ICSR del 4 % es comparable al perfil de seguridad del producto de referencia [ 42 ].
2.1.3 Rixathon®/Riximyo®/ Arasamila® (Sandoz)
GP2013 (Rixathon® /Riximyo®/ Arasamila®) es un biosimilar de rituximab desarrollado por Sandoz en Austria. GP2013 fue registrado en Argentina por Novartis con la marca Rixathon®. GP2013 fue registrado por Sandoz en Brasil como Riximyo® y también fue registrado por Sandoz en México con la marca Arasamila®. Ha estado en uso clínico para el tratamiento de pacientes con NHL, CLL, artritis reumatoide y otras condiciones autoinmunes [ 43 ].
Según una comparabilidad fisicoquímica y funcional con el rituximab de referencia, se demostró que la secuencia de aminoácidos y la masa molecular de GP2013 eran idénticas [ 44 ]. Además, las modificaciones específicas de aminoácidos y el patrón de glicanos no se distinguían del rituximab original [ 44 ]. Los bioensayos y los ensayos de unión para medir la funcionalidad revelaron un resultado similar para el biosimilar y el anticuerpo de referencia, especialmente la potencia de ADCC, que se probóin vitroyen vivo[ 44 , 45 ]. El ejercicio de comparabilidad preclínica realizado en monos cynomolgus reveló que la farmacocinética y la farmacodinámica eran comparables entre GP2013 y el rituximab de referencia [ 45 ].
Se realizó un estudio clínico aleatorizado doble ciego en el que los pacientes con artritis reumatoide con respuesta inadecuada o intolerancia al tratamiento con factor de necrosis tumoral-α (TNFα) recibieron GP2013 o rituximab de referencia junto con metotrexato y ácido fólico [ 46 ]. En este ensayo clínico, los perfiles de eficacia, seguridad e inmunogenicidad fueron similares entre GP2013 y el original rituximab, además de la equivalencia mostrada en los parámetros farmacocinéticos y farmacodinámicos [ 46 ].
Se realizaron estudios adicionales de eficacia, seguridad, tolerabilidad, farmacocinética y farmacodinámica de GP2013 más ciclofosfamida, vincristina y prednisona (GP2013-CVP) en comparación con rituximab de referencia en un ensayo clínico multinacional, doble ciego, aleatorizado, de fase III en adultos con , linfoma folicular en estadio avanzado [ 47 ]. Se observó equivalencia de la respuesta global en el grupo con GP2013 (87 %) en comparación con el rituximab de referencia (88 %) [ 47]. En función de los resultados de eficacia primarios y secundarios, se determinó la equivalencia entre GP2013 y el rituximab de referencia en términos de tasa de respuesta general para la evaluación del tumor, y respuesta completa, respuesta parcial, enfermedad estable y enfermedad progresiva similares, en pacientes con linfoma folicular en estadio avanzado no tratado. demostrado [ 47 ].
Las frecuencias generales de eventos adversos comunes y eventos adversos graves fueron comparables entre ambos grupos de tratamiento en LNH folicular (fases de combinación y mantenimiento) y en artritis reumatoide. Los perfiles de seguridad, incluida la inmunogenicidad de GP2013 en las poblaciones fundamentales, son coherentes con el perfil de seguridad conocido del medicamento de referencia notificado en los ensayos clínicos y la vigilancia posterior a la comercialización. Además, no se detectaron riesgos de seguridad en pacientes que cambiaron del medicamento de referencia a GP2013 [ 47 ].
2.1.4 Truxima® (Celltrion)
CT-P10 (Truxima®) es un biosimilar de rituximab desarrollado por Celltrion Healthcare en Corea del Sur. CT-P10 es comercializado con la marca Truxima® por Celltrion en Brasil y Colombia y por Saval en Chile. Truxima® fue aprobado para el tratamiento de LNH, CLL, artritis reumatoide y granulomatosis con poliangeítis y poliangeítis microscópica [ 48 , 49 ].
CT-P10 ha mostrado una gran similitud en su estructura primaria, estructuras de orden superior, modificaciones postraduccionales y actividades biológicas [ 50 ]. La biosimilitud de CT-P10 con el rituximab de referencia se logró con una evaluación de similitud de 3 vías realizada entre CT-P10, EU-rituximab y US-rituximab, centrándose en los atributos de calidad fisicoquímicos y biológicos [ 50 ]. Una multitud de análisis reveló que CT-P10 tiene estructuras primarias y de orden superior idénticas en comparación con el producto original. También se encontró que los perfiles de pureza/impureza de CT-P10 medidos por los niveles de agregados, fragmentos, forma no glicosilada e impurezas relacionadas con el proceso son comparables con los del medicamento de referencia [ 50]. En cuanto a la modificación postraduccional, CT-P10 contiene una variante de piroglutamato N-terminal ligeramente menor, que se sabe que no afecta la eficacia ni la seguridad del producto. Las matrices de ensayos biológicos representativos de los mecanismos de acción conocidos y putativos de rituximab han demostrado que las actividades biológicas de CT-P10 están dentro del rango de calidad del rituximab de referencia [ 50 ].
Se realizó un ensayo clínico de Fase I para evaluar la farmacocinética de CT-P10 y el rituximab de referencia. Los resultados del estudio demostraron que CT-P10 y el rituximab de referencia fueron estadísticamente equivalentes después de un solo ciclo de tratamiento en la semana 24. El estudio también encontró que la eficacia, la farmacodinámica, la inmunogenicidad y la seguridad fueron similares hasta en dos ciclos de tratamientos de hasta 72 semanas. [ 51 ]. Los resultados de otro estudio clínico de extensión de etiqueta abierta de Fase I demostraron que cambiar a CT-P10 desde el rituximab de referencia fue efectivo con una seguridad comparable a continuar con CT-P10 durante dos años [ 52 ].
Se llevaron a cabo ensayos clínicos comparativos de fase III sobre CT-P10 en pacientes con artritis reumatoide, linfoma folicular avanzado y linfoma folicular de baja carga tumoral (LTBFL) [ 52 , 53 ]. Los resultados mostraron que el tratamiento con CT-P10 en pacientes con artritis reumatoide resultó en perfiles de eficacia, PK, PD, inmunogenicidad y seguridad muy similares en comparación con los tratados con rituximab de referencia [ 52 ]. CT-P10 también demostró ser equivalente en términos de eficacia y seguridad en pacientes con LTBFL [ 53 ].
2.1.5 Zaytux® (AryoGen)
Zaytux®/Zytux® es un biosimilar de rituximab desarrollado por AryoGen Biopharma en Irán y distribuido en Perú por Perulab. Se utiliza para el tratamiento de pacientes adultos con LNH, LLC, artritis reumatoide (AR) y granulomatosis con poliangeítis (GPA) y poliangeítis microscópica (MPA).
Los estudios de comparabilidad revelaron propiedades fisicoquímicas y biológicas similares entre Zytux y el rituximab de referencia [ 54 ]. Se encontraron estructuras primarias y modificaciones postraduccionales similares. Además, se obtuvieron estructuras secundarias, terciarias y cuaternarias comparables para el original y el biosimilar de rituximab, analizadas mediante espectroscopia de CD, espectroscopia de RMN, FTIR y MS de movilidad de iones. Se encontraron pequeñas diferencias en los estudios de determinación de masa en el biosimilar Zytux® con respecto al rituximab de referencia; los más relevantes son el truncamiento incompleto de la lisina C-terminal de las cadenas pesadas y una diferencia de 2 Da en las cadenas ligeras. Los datos han mostrado una gran similitud en el patrón de N-glucanos y la identidad de las principales glicoformas [ 54]. La evaluación de comparabilidad de lote a lote de los N-glucanos liberados de rituximab y su biosimilar mostró que sus patrones de N-glucanos son cualitativamente similares, pero cuantitativamente heterogéneos, aunque se consideran cambios aceptables. Los estudios de unión por resonancia de plasmones de superficie mostraron que la unión de Fc de Zytux y rituximab al receptor Fc humano recombinante y las variantes del receptor FcRn exhiben valores de constante de equilibrio (KD) similares. Además, se obtuvieron resultados comparables a partir de ensayos de unión a C1q y ensayos de citotoxicidad dependiente del complemento (CDC). Además, se realizaron ensayos de unión entre los anticuerpos y CD20 y mostraron afinidades similares [ 54 ].
Los datos de los ensayos clínicos en pacientes con CLL y NHL mostraron resultados comparables en términos de eficacia y seguridad para Zytux® y el rituximab de referencia [ 55 ]. Los pacientes con LLC se incluyeron en un estudio aleatorio doble ciego que mostró resultados no inferiores y comparables en términos de eficacia (tasa de respuesta general y marcadores específicos de células B) y seguridad (reacciones a la infusión, toxicidad hematológica y toxicidad no hematológica). Otro estudio realizado en 10 pacientes con LLC y 10 con LNH evaluó la seguridad y eficacia de Zytux® en comparación con el rituximab de referencia [ 56 ], concluyendo que Zytux® no era inferior al rituximab de referencia, y era comparable e incluso mejor en términos de seguridad y eficacia.
2.2 Trastuzumab
Desarrollado por Genentech en los Estados Unidos, el trastuzumab se comercializa con la marca Herceptin® y es fabricado por Roche. Aprobado por la FDA en 1998, trastuzumab fue el primer anticuerpo monoclonal humanizado contra el cáncer. Es un anticuerpo monoclonal IgG1 / κ humanizado que se dirige al dominio extracelular del receptor 2 del factor de crecimiento epidérmico humano (HER2) y se usa para el tratamiento del cáncer de mama metastásico o temprano positivo para HER2 [ 57 ]. En 2021, hay un total de cuatro biosimilares de trastuzumab diferentes aprobados en América Latina, con siete marcas diferentes comercializadas en Argentina, Brasil, Colombia y Perú ( Tabla 1 ).
2.2.1 Kanjinti® (Amgen)
ABP 980 (Kanjinti®) es un biosimilar de trastuzumab desarrollado en los Estados Unidos por Amgen. ABP 980 fue aprobado por la FDA para todas las indicaciones aprobadas del producto de referencia, incluido el tratamiento del cáncer de mama metastásico y adyuvante con sobreexpresión de HER2 y el adenocarcinoma gástrico o de la unión gastroesofágica metastásico con sobreexpresión de HER2. Fue registrado en Argentina y Perú como Kanjinti® por Varifarma SA
Se demostró que ABP 980 tiene propiedades fisicoquímicas y funcionales similares a las del trastuzumab de referencia, la fisicoquímica es similar al trastuzumab de referencia en términos de estructura primaria y de orden superior, estructura de carbohidratos, propiedades de unión cinética (vs. trastuzumab) y pureza [ 58 , 59 , 60 ]. Las diferencias menores entre los dos agentes no se consideraron clínicamente significativas.
En un estudio clínico de dosis única, se demostró la similitud farmacocinética de ABP 980 con el trastuzumab de EE. UU. y la UE. No se observaron diferencias en la seguridad y la tolerabilidad entre los tratamientos y ningún sujeto dio positivo para la unión de anticuerpos [ 61 ]. Además, se demostró que la farmacodinámica tiene una potencia similar a la del trastuzumab de referencia procedente de la UE en términos de inhibición de la proliferación e inducción de ADCC [ 59 ].
En el estudio clínico de fase III LILAC, ABP 980 demostró una eficacia clínica y una tolerabilidad similares a las del trastuzumab de referencia en pacientes con cáncer de mama temprano positivo para HER2 [ 62 , 63 ]. Además, los perfiles de inmunogenicidad y seguridad de ABP 980 fueron similares a los del trastuzumab de referencia, y un solo cambio de trastuzumab de referencia a ABP 980 no tuvo impacto en la inmunogenicidad o seguridad de ABP 980 [ 62 ]. Cambiar de trastuzumab a ABP 980 no tuvo un impacto significativo en la supervivencia libre de eventos y no afectó negativamente su tolerabilidad [ 63]. Los análisis de sensibilidad se llevaron a cabo en base a la evaluación del laboratorio central de muestras de tumores; las estimaciones para los dos medicamentos estaban contenidas dentro de los márgenes de equivalencia predefinidos, lo que indica una eficacia similar [ 62 ]. ABP 980 y el trastuzumab de referencia tuvieron resultados de seguridad similares en las fases neoadyuvante y adyuvante del estudio [ 62 , 64 ].
2.2.2 Tuzepta® / Zedora®/ Ogivri®/ Bisinte® (Biocon)
Conocido como MYL-1401O (Tuzepta®, Zedora®, Ogivrí®y Bisintex®), este biosimilar de trastuzumab fue desarrollado en India por Biocon/Mylan. Fue aprobado por la FDA en 2017 y en Estados Unidos lo comercializa Mylan con la marca Ogivri®. MYL-1401O está indicado para el tratamiento del cáncer de mama con sobreexpresión de HER2 en pacientes que han recibido uno o más regímenes de quimioterapia para la enfermedad metastásica. En Argentina, MYL-1401O fue registrado como Tuzepta® por Laboratorio Raffo; en Brasil fue registrado como Zedora® por Libbs; en Colombia fue registrado como Ogivri® y distribuido por Mylan. En Bolivia, Chile, Paraguay y Perú se comercializa como Bisintex® y es distribuido por PharmaTech Boliviana en Bolivia, Recalcine en Chile, Pharma International en Paraguay y Abbott en Perú.
La totalidad de la evidencia de MYL-1401O respalda su biosimilitud con el trastuzumab de referencia en base a un ejercicio de comparabilidad, que incluye evaluaciones de similitud analítica estructural y funcional y un estudio clínico confirmatorio [ 65 , 66]. Los estudios de comparabilidad realizados para MYL-1401O y trastuzumab de referencia incluyeron un estudio de estabilidad fisicoquímica, donde se probaron todas las condiciones de almacenamiento. Los resultados mostraron que no hubo cambios en la estructura terciaria de MYL-1401O según lo evaluado por ultravioleta de segundo derivado y análisis espectral derivado de fluorescencia, y no se observó evidencia de formación o fragmentación de oligómeros según lo evaluado por cromatografía de exclusión en gel y dispersión de luz dinámica. . La cromatografía de intercambio iónico no mostró cambios significativos en la distribución de variantes iónicas [ 67 ].
MYL-1401O fue bien tolerado y demostró perfiles farmacocinéticos y de seguridad similares al trastuzumab de referencia en voluntarios sanos [ 68 ]. Esto se demostró con un estudio de fase I de un solo centro, aleatorizado, doble ciego, de tres brazos, de grupos paralelos realizado en voluntarios varones adultos sanos que recibieron MYL-1401O o trastuzumab de referencia como una infusión intravenosa de 90 minutos. El estudio clínico demostró que entre las mujeres con cáncer de mama metastásico positivo para HER2 que recibieron taxanos, el uso de MYL-1401O en comparación con el trastuzumab de referencia resultó en una tasa de respuesta general equivalente a las 24 semanas [ 66 ].
2.2.3 Trazimera® (Pfizer)
PF-05280014 (Trazimera®) es un biosimilar de trastuzumab desarrollado en los Estados Unidos por Pfizer y aprobado en la Unión Europea en 2018 [ 69 ]. Está indicado para el tratamiento de pacientes adultos con cáncer de mama y gástrico metastásico HER2 positivo [ 70 ]. Trazimera® fue registrada en Argentina por Pfizer y en Brasil por Wyeth.
Se demostró que la caracterización fisicoquímica es similar al trastuzumab de referencia (tanto de la UE como de los EE. UU.) en términos de estructuras primarias, secundarias y terciarias, modificaciones postraduccionales, variantes de carga, pureza y estabilidad [ 71 ]. No se encontraron diferencias clínicamente significativas entre PF-05280014 y el trastuzumab de referencia de la UE y los EE. UU. después de los cambios de formulación (es decir, un ligero cambio en la fucosilación a total, la galactosilación terminal y las especies G0). Las diferencias estructurales y funcionales menores entre PF-05280014 y el trastuzumab de referencia no se consideraron clínicamente relevantes [ 71 ].
Se encontró que las propiedades farmacodinámicas de PF-05280014 son similares a las del trastuzumab de referencia (tanto de la UE como de los EE. UU.) en términos de actividad biológica, incluidas las características funcionales y de unión (p. funciones basadas, actividades ADCC y ADCP). Se demostró una eficacia equivalente y una tolerabilidad similar al trastuzumab de referencia en el cáncer de mama HER2 positivo metastásico, y una eficacia y tolerabilidad similares al trastuzumab de referencia en mujeres con cáncer de mama HER2 positivo temprano [ 69 ].
Se llevaron a cabo varios estudios farmacocinéticos que demostraron la similitud entre PF-05280014 y trastuzumab-EU en términos de actividad farmacocinética. Uno de estos estudios se realizó en un ensayo clínico multinacional, doble ciego, aleatorizado y comparativo de PF-05280014 frente a trastuzumab-EU, en el que un total de 702 pacientes con cáncer de mama metastásico fueron tratados con PF-05280014 y trastuzumab-EU. PF-05280014 y trastuzumab-EU tenían parámetros farmacocinéticos similares y covariables farmacocinéticas influyentes en pacientes con cáncer de mama metastásico positivo para HER2 [ 72 ]. Finalmente, otro estudio aleatorizado, doble ciego [ 71], comparó la farmacocinética, la eficacia, la seguridad y la inmunogenicidad de PF-05280014 y el producto de referencia trastuzumab como tratamiento neoadyuvante para el cáncer de mama HER2 positivo operable. PF-05280014 demostró una farmacocinética no inferior y una eficacia, seguridad e inmunogenicidad comparables a trastuzumab-EU en pacientes con cáncer de mama HER2 positivo operable que reciben quimioterapia neoadyuvante [ 71 ].
Se informaron resultados adicionales sobre seguridad, eficacia, inmunogenicidad y supervivencia general de pacientes con cáncer de mama metastásico positivo para HER2 en un estudio aleatorizado, doble ciego, que comparó PF-05280014 con trastuzumab de referencia cuando a cada paciente se le administró paclitaxel como tratamiento de primera línea [ 73 ] . El estudio no mostró diferencias destacables entre ambos grupos en la supervivencia libre de progresión ni en la supervivencia global. Los resultados de seguridad y la inmunogenicidad fueron similares entre los grupos de tratamiento. Además, cuando se administró como tratamiento de primera línea para el cáncer de mama metastásico positivo para HER2, se demostró la equivalencia de PF-05280014 más paclitaxel con trastuzumab-EU más paclitaxel en términos de tasa de respuesta objetiva.
2.2.4 Herzuma (Celltrion)
CT-P6 (Herzuma®) es un biosimilar de trastuzumab desarrollado en Corea del Sur por Celltrion. CT-P6 es un antagonista del receptor HER2 aprobado en la Unión Europea para el tratamiento del cáncer de mama con sobreexpresión de HER2. Fue registrado en Brasil por Celltrion con la marca Herzuma®.
Los estudios de comparabilidad que evaluaron las similitudes analíticas entre CT-P6 y el trastuzumab de referencia demostraron que exhibe propiedades estructurales y fisicoquímicas muy similares, así como actividades ADCC y antiproliferativas, en comparación con el trastuzumab de referencia [ 74 ]. Con respecto a la glicosilación, los glucanos galactosilados, el ácido siálico y las glucaciones, la comparación entre CT-P6 y los productos de referencia trastuzumab mostró que, aunque se detectaron variabilidades significativas en CT-P6, estaban en el mismo rango que las observadas en el producto de referencia [ 74 ].
La comparabilidad clínica entre CT-P6 y el trastuzumab de referencia se probó en un estudio aleatorizado, doble ciego, de dos grupos, de grupos paralelos y de dosis única para evaluar la farmacocinética, la seguridad y la inmunogenicidad de CT-P6 en comparación con el trastuzumab de referencia en personas sanas. temas [ 75 ]. En este estudio, se observó equivalencia entre condiciones, con concentraciones séricas similares en el período analizado, perfiles de seguridad similares, sin eventos adversos graves o muertes, y ningún sujeto dio positivo para anticuerpos antidrogas [ 75 ].
Otros estudios incluyeron un estudio de fase III, doble ciego, aleatorizado, de grupos paralelos con control activo, multicéntrico, internacional y prospectivo para comparar la eficacia y la seguridad de CT-P6 y el trastuzumab de referencia como tratamiento neoadyuvante y adyuvante en pacientes con cáncer de mama en estadio temprano. cáncer HER2 positivo. Este ensayo demostró que el CT-P6 neoadyuvante tenía una eficacia comparable al trastuzumab de referencia y confirmó la similitud en la seguridad, incluido el riesgo comparable de cardiotoxicidad. Cuando se usó como terapia adyuvante después del tratamiento neoadyuvante, CT-P6 demostró comparabilidad con el trastuzumab de referencia en términos de prevención de la enfermedad progresiva en pacientes con cáncer de mama en estadio temprano positivo para HER2 [ 76 ].
Actualmente, CT-P6 está indicado para el tratamiento de pacientes con cáncer de mama metastásico que tienen tumores que sobreexpresan HER2, para el tratamiento de pacientes que ya han recibido tratamientos de quimioterapia para sus enfermedades metastásicas, en combinación con paclitaxel o docetaxel para el tratamiento de pacientes que aún no han recibido quimioterapia. CT-P6 en combinación con capecitabina intravenosa o 5-fluorouracilo (5-FU) y un agente de platino está indicado para el tratamiento de pacientes con adenocarcinoma de estómago o unión gastroesofágica HER2 positivo inoperable, localmente avanzado, recurrente o metastásico, que tienen no recibió tratamiento previo para el cáncer metastásico [ 77 ].
2.3 Infliximab
Infliximab fue desarrollado en los Estados Unidos por Janssen Biotech, aprobado por la FDA en 1998 y comercializado bajo la marca Remicade®. Es un anticuerpo monoclonal recombinante quimérico con isotipo IgG1/κ que se dirige al TNFα y fue el primer inhibidor del TNFα utilizado para tratar la inflamación crónica [ 78 ]. Se utiliza para el tratamiento de varias afecciones, incluida la enfermedad inflamatoria intestinal (EII), la enfermedad de Crohn, la colitis ulcerosa, la artritis reumatoide, la espondilitis anquilosante, la psoriasis, la artritis psoriásica y la enfermedad de Behçet. Hay dos biosimilares de infliximab aprobados en América Latina, con amplia distribución en la región, comercializados bajo cuatro marcas diferentes en Argentina, Brasil, Chile, Colombia, Ecuador, México, Paraguay y Perú ( Tabla 1 ).
2.3.1 Remsima®/Flixceli® (Celltrion)
CT-P13 (Remsima®/Flixceli®) es un biosimilar de infliximab desarrollado en Corea del Sur por Celltrion. Fue el primer anticuerpo monoclonal biosimilar aprobado por la Unión Europea [ 12]. CT-P13 está indicado para el tratamiento de la artritis reumatoide, la espondilitis anquilosante con ensayos clínicos, y por extrapolación para el tratamiento de la artritis psoriásica y la psoriasis. También está indicado por extrapolación para adultos y niños mayores de 6 años para enfermedad de Crohn y colitis ulcerosa. En Argentina, el CT-P13 fue registrado por Gobbi-Novag con la marca Remsima®. En Brasil, Colombia, México y Paraguay CT-P13 fue registrado por Celltrion como Remsima®. En Chile, CT-P13 fue registrado por Saval como Remsima®. En Ecuador, CT-P13 fue registrado por Oxialfarm también como Remsima®. En Perú, CT-P13 se comercializa como Flixceli® y fue registrado por AC Pharma.
Las propiedades fisicoquímicas y biológicas de CT-P13 se han caracterizado ampliamente en comparación con las del infliximab de referencia, lo que demuestra una gran similitud en sus propiedades fisicoquímicas en comparación con el original [ 79 ]. Entre las propiedades que se evaluaron están la estructura primaria y los principales órdenes de estructura, tipo y distribución de glicanos, purezas/impurezas, número y distribución de variantes cargadas, unión al objetivo molecular y potencia biológica. También se ha demostrado una actividad similar en farmacodinámica [ 80 ], donde se ha demostrado que ambos tienen afinidades de unión equivalentes a TNFα y falta de unión a TNFβ y TNFα de otras especies.in vitrolos estudios demostraron efectos apoptóticos equivalentes y citotoxicidad mediada por células dependientes de anticuerpos (ADCC) y CDC, así como reactividad cruzada similar en tejido humano [ 81 ].
Se llevaron a cabo estudios clínicos para demostrar la equivalencia entre CT-P13 e infliximab de referencia en términos de PK/PD, seguridad y eficacia en pacientes con artritis reumatoide y espondilitis anquilosante activa [ 80 , 82 ]. Además, se realizaron estudios clínicos en pacientes con colitis ulcerosa y enfermedad de Crohn, en los que también se ha observado comparabilidad con el infliximab de referencia en términos de eficacia y seguridad, lo que también proporciona evidencia de intercambiabilidad entre ambos [ 79 , 83]. Se ha visto más evidencia de intercambiabilidad tras el cambio de tratamiento de referencia infliximab a CT-P13 en pacientes con artritis reumatoide y espondilitis anquilosante, ya que es bien tolerado y los resultados son comparables en términos de eficacia, inmunogenicidad y seguridad [ 80 , 82 , 83 , 84 ].
2.3.2 Ixifi®/Xilfya® (Pfizer)
Otro biosimilar de infliximab es PF-06438179/GP1111 (Ixifi® / Xilfya®), que fue desarrollado en los Estados Unidos por Pfizer. PF-06438179 fue aprobado por la FDA en 2017 como tratamiento para pacientes con artritis reumatoide, enfermedad de Crohn, enfermedad de Crohn pediátrica, colitis ulcerosa, espondilitis anquilosante, artritis psoriásica y psoriasis en placas. PF-06438179 fue registrado en Argentina con la marca Ixifi® por Pfizer. En Brasil, fue registrado como Xilfya® por Wieth.
Los estudios de comparabilidad no clínica entre PF-06438179 y el infliximab de referencia han mostrado una estructura proteica similar, con perfiles de mapas de péptidos superponibles y las mismas masas de péptidos, lo que indica secuencias de aminoácidos idénticas. Además, los datos sobre las modificaciones postraduccionales, las propiedades bioquímicas y la función biológica brindaron un fuerte apoyo a la similitud no clínica de PF-06438179 [ 85 ].
Los estudios clínicos que compararon la farmacocinética, la seguridad y la inmunogenicidad de PF-06438179 y el infliximab de referencia incluyeron un estudio de administración intravenosa de dosis única en pacientes adultos sanos, de tres brazos, doble ciego, aleatorizado (1:1:1) con grupos paralelos. Los resultados farmacocinéticos obtenidos en estudios con pacientes sanos mostraron perfiles similares de concentraciones séricas-tiempo en todos los grupos de tratamiento. Los eventos adversos fueron similares entre PF-06438179 y el infliximab de referencia y los perfiles de anticuerpos neutralizantes y antidrogas fueron similares entre los grupos [ 86 ].
La comparabilidad clínica de PF-06438179 con el infliximab de referencia también se probó en un estudio controlado en pacientes con artritis reumatoide con una respuesta inadecuada al metotrexato. Los resultados no muestran diferencias clínicamente significativas en eficacia, farmacodinamia, inmunogenicidad y seguridad entre los pacientes que recibieron PF-06438179 e infliximab de referencia y en pacientes que hicieron la transición (intercambio único) de infliximab de referencia a PF-06438179 [ 87 ].
2.4 Adalimumab
Desarrollado en los Estados Unidos por Abbott (hoy AbbVie), Adalimumab (Humira®) fue el primer anticuerpo monoclonal completamente humano aprobado por la FDA en 2002. Adalimumab fue aprobado en 2003 en la Unión Europea con las marcas Humira® y Trudexa®. Es un anticuerpo monoclonal IgG1/κ anti-factor de necrosis tumoral α (anti-TNFα) totalmente humano que previene la interacción del TNFα con sus receptores, interfiriendo así con la señalización inflamatoria central de las enfermedades autoinmunes crónicas como la artritis reumatoide, la artritis psoriásica, espondilitis anquilosante, enfermedad de Crohn, enfermedad de Crohn pediátrica, psoriasis crónica de moderada a grave y artritis idiopática juvenil [ 88]. Actualmente hay tres biosimilares de adalimumab aprobados en América Latina, comercializados bajo tres marcas en Argentina, Brasil y Perú ( Tabla 1 ).
2.4.1 Amgevita® (Amgen)
ABP 501 (Amgevita®/ Amjevita®) es un biosimilar de adalimumab desarrollado en los Estados Unidos por Amgen. Fue el primer biosimilar de adalimumab aprobado por la FDA en 2016 y por la EMA en 2017 [ 69 ]. Está autorizado para el tratamiento de enfermedades inflamatorias en adultos, incluida la artritis reumatoide de moderada a grave; artritis psoriásica; espondilitis anquilosante activa grave; espondiloartritis axial grave; psoriasis crónica en placas; hidradenitis supurativa; intermedia no infecciosa, posterior y panuveítis; Enfermedad de Crohn y colitis ulcerosa. ABP 501 fue registrado en Argentina, Brasil y Colombia por Amgen con la marca Amgevita®. En Perú, ABP 501 fue registrado por TecnoFarma también con la marca Amgevita®.
ABP 501 es un anticuerpo monoclonal recombinante completamente humano con la misma secuencia de aminoácidos, forma farmacéutica y concentración de dosificación que el adalimumab de referencia. Sin embargo, no está formulado con los mismos excipientes que adalimumab e incluye diferentes componentes tampón y estabilizadores; debido a esto, se han realizado varios estudios de similitud entre ellos. Se ha demostrado que ABP 501 es tanto analítica como funcionalmente similar al adalimumab de referencia [ 89 , 90 ]. Los resultados de los estudios analíticos que evaluaron la identidad, las propiedades generales, la estructura primaria y de orden superior, la estructura de los carbohidratos, el perfil isoeléctrico, la pureza y las impurezas, y los perfiles de degradación térmica forzada han confirmado que ABP 501 es estructuralmente similar al adalimumab de referencia [ 89 ].]. Además, los resultados de los estudios de caracterización funcional han demostrado que ABP 501 y el adalimumab de referencia tienen una afinidad de unión similar al TNFα y una inhibición comparable de las actividades del TNFα.in vitro. Además, ABP 501 y el adalimumab de referencia han mostrado una inducción comparable de funciones efectoras y también han demostrado ser similares a adalimumab con respecto a la unión a un panel de receptores Fc, incluidos FcγRIa, FcγRIIa, FcγRIIIa (158V), FcγRIIIa (158F) y FcRn [ 90 ].
En cuanto a la farmacocinética, se realizó un estudio clínico en adultos sanos que recibieron ABP 501 o adalimumab de referencia [ 91 ]. Los resultados del estudio mostraron que no hubo diferencias significativas entre ABP 501 y el adalimumab de referencia en términos de seguridad, eficacia e inmunogenicidad en las condiciones de uso aprobadas para adalimumab y de acuerdo con las normas y directrices para el desarrollo de biosimilares [ 91 ]. Los estudios clínicos de fase III han demostrado que ABP 501 y el adalimumab de referencia tienen perfiles similares de eficacia clínica, seguridad e inmunogenicidad durante 52 semanas de tratamiento en una población sensible de pacientes inmunocompetentes con psoriasis [ 92]. Además, los datos de un estudio de equivalencia de fase III aleatorizado, doble ciego y diferente en pacientes con artritis reumatoide de moderada a grave indicaron que la eficacia clínica, la seguridad y la inmunogenicidad de ABP 501 son similares a las del adalimumab de referencia [ 93 ]
2.4.2 Hyrimoz® (Sandoz)
El biosimilar de adalimumab GP2017 (Hyrimoz®) fue desarrollado en Alemania por Sandoz y en 2018 fue autorizado en la Unión Europea para su uso en pacientes con artritis reumatoide, psoriasis en placas, enfermedad de Crohn, uveítis y colitis ulcerosa y todas las indicaciones para las que está aprobado el adalimumab de referencia [ 94 ]. GP2017 fue registrado en Brasil por Sandoz con la marca Hyrimoz®.
Se ha demostrado que GP2017 exhibe similitud con el adalimumab de referencia con respecto a las estructuras primarias, secundarias y terciarias, la estructura de carbohidratos, el tamaño molecular, las cargas y las impurezas. Las diferencias entre GP2017 y el adalimumab de referencia en las variantes de glicosilación no fueron clínicamente relevantes [ 94 ]. También se determinó la similitud en las determinaciones de la actividad funcional de la unión a TNFα, a los subtipos de receptores Fcγ humanos ya FcRn. Otros estudios comparativos funcionales incluyen CDC, ADCC, C1q, inhibición de la apoptosis e inducción de la apoptosis/señalización inversa [ 95 ]
Se realizó un estudio clínico de comparabilidad de GP2017 con adalimumab de referencia para evaluar la similitud en la farmacocinética, la seguridad y la inmunogenicidad durante 72 días después de la inyección [ 96 ]. En el estudio, la concentración sérica máxima y el AUC desde el momento de la dosificación extrapolados al infinito se observaron dentro del margen de similitud predeterminado entre GP2017 y el adalimumab de referencia. La mayoría de los eventos adversos emergentes del tratamiento fueron de intensidad leve o moderada y la determinación de anticuerpos antidrogas fue similar entre los grupos, con un 57,9 % de GP2017, un 69,8 % para EU-adalimumab y un 69,5 % para US-adalimumab [ 96 ].
Además, la eficacia clínica de GP2017 en comparación con el adalimumab de referencia se probó en un estudio aleatorizado de fase III en psoriasis en pacientes con psoriasis en placas de moderada a grave o artritis reumatoide. En este estudio, se demostró que los perfiles de tolerabilidad, seguridad e inmunogenicidad de los dos agentes eran similares. Se mostró la eficacia entre los grupos, donde el cambio múltiple entre GP2017 y adalimumab de referencia (hasta cuatro veces) no tuvo impacto en la eficacia, tolerabilidad o inmunogenicidad. El papel del adalimumab de referencia en el tratamiento de las afecciones inflamatorias autoinmunes está bien establecido y este estudio proporciona evidencia de que GP2017 es una alternativa biosimilar eficaz para los pacientes que requieren terapia con adalimumab [ 97 ].
2.4.3 Xilbrilada® (Pfizer)
PF-06410293 (Abrilada®/Xilbrilada®) es un biosimilar de adalimumab desarrollado en Estados Unidos por Pfizer y aprobado por la FDA en 2019, donde se comercializa con la marca Abrilada®. PF-06410293 está indicado para el tratamiento de pacientes con artritis reumatoide, artritis psoriásica, espondilitis anquilosante, enfermedad de Crohn del adulto, colitis ulcerosa, psoriasis en placas y artritis idiopática juvenil. PF-06410293 fue registrado en Brasil por Wyeth, donde se comercializa como Xilbrilada®.
Se realizaron estudios no clínicos comparativos entre PF-06410293 y adalimumab de referencia y confirmaron la similitud [ 98 ]. El análisis estructural que evaluó el mapeo de péptidos mostró perfiles cromatográficos similares, lo que confirma que las secuencias de aminoácidos PF-06410293 y el adalimumab de referencia son idénticos. Los datos sobre las modificaciones postraduccionales, las propiedades bioquímicas y la función biológica brindaron un fuerte respaldo a la similitud analítica. La unión a TNFα fue similar para PF-06410293 y adalimumab de referencia. Además,en vivoLos estudios en ratas mostraron que la aplicación intravenosa de PF-06410293 y el adalimumab de referencia fueron bien tolerados y exhibieron una farmacocinética similar, con una concentración máxima del fármaco y un AUC equivalentes [ 98 ].
La similitud clínica entre PF-06410293 y el adalimumab de referencia se probó en un estudio clínico, doble ciego, aleatorizado, comparativo, de eficacia en individuos con artritis reumatoide gravemente activa y con respuesta inadecuada al metotrexato. El estudio demostró equivalencia terapéutica (similitud) y respuestas similares entre tratamientos con PF-06410293 y adalimumab de referencia. El estudio muestra la ausencia de diferencias clínicamente significativas en eficacia, farmacodinamia, inmunogenicidad y seguridad entre los individuos que recibieron PF-06410293 o adalimumab de referencia. Además, se observó una respuesta equivalente en pacientes que pasaron de PF-06410293 a adalimumab de referencia y en aquellos que pasaron de adalimumab de referencia a PF-06410293.99 ].
2.5 Bevacizumab
Bevacizumab (Avastin®) es un anticuerpo monoclonal humanizado con isotipo IgG1/κ que se dirige al factor de crecimiento vascular-endotelial (VEGF), que a su vez previene la proliferación endotelial e inhibe la angiogénesis. Fue desarrollado por Genentech, recibiendo su primera aprobación en Estados Unidos en 2004 por parte de la FDA, y actualmente lo comercializa Roche. Originalmente indicado en uso combinado con quimioterapia estándar contra el cáncer de colon metastásico, desde entonces ha sido aprobado para su uso en ciertos cánceres de pulmón, cánceres renales, cánceres de ovario y glioblastoma multiforme del cerebro. Dos biosimilares de bevacizumab se comercializan en América Latina para 2021. Están aprobados en Argentina, Brasil, Colombia, Ecuador y Paraguay, y se comercializan bajo dos marcas ( Tabla 1 ).
2.5.1 Bevax® (mAbxience)
BEVZ92 (Bevax®) es un anticuerpo biosimilar de bevacizumab desarrollado en Argentina por PharmaADN (hoy mAbxience) y comercializado por Laboratorios Elea en Argentina. Está indicado en combinación con otros agentes quimioterapéuticos y biológicos para el cáncer metastásico de colon [ 100 ], y por extrapolación a adultos con cáncer metastásico de recto, mama, riñón, glioblastoma, ovario, peritoneo, útero y cáncer de pulmón no microcítico. . BEVZ92 es distribuido por Grünenthal en Ecuador y distribuido por Laoratorios Bioéticos en Paraguay con la marca Bevax®.
La comparabilidad clínica de BEVZ92 y el bevacizumab de referencia se realizó en el ensayo clínico NCT02069704, que finalizó en junio de 2017. Este fue un estudio multicéntrico, abierto, de bioequivalencia de BEVZ92 y el bevacizumab de referencia, aleatorizado con 2 brazos paralelos para comparar la eficacia. , seguridad, inmunogenicidad y perfil farmacocinético de BEVZ92 y bevacizumab de referencia en combinación con quimioterapia para el cáncer colorrectal metastásico [ 100 ]. Los pacientes han mostrado similitudes en la farmacocinética comparando la relación media geométrica del AUC en pacientes tratados con BEVZ92 y con bevacizumab de referencia [ 100]. Además, la respuesta objetiva, el beneficio clínico y la supervivencia libre de progresión fueron similares para los grupos BEVZ92 y bevacizumab de referencia. El perfil de seguridad no mostró diferencias relevantes entre ambos brazos del estudio, con niveles similares de eventos adversos de grado 3 o 4 y efectos adversos graves [ 100]. La inmunogenicidad evaluada como la incidencia de anticuerpos antidrogas fue similar y baja para ambos brazos del estudio. Los resultados informados muestran que, cuando se usan de la misma manera, BEVZ92 y el bevacizumab de referencia son muy similares en cuanto a farmacocinética, inmunogenicidad, seguridad y eficacia para el tratamiento del cáncer colorrectal metastásico. Romera et al. también informaron que BEVZ92 era similar al bevacizumab de referencia en una extensa caracterización fisicoquímica y funcional, incluida la estructura primaria, la estructura de orden superior, la actividad biológica y la afinidad de unión a VEGF, aunque no se mostraron los datos [ 100 ].
En 2016 se estableció un Registro de Tratamiento para la farmacovigilancia de BEVZ92 para recoger las reacciones adversas a medicamentos (RAM) de los pacientes tratados con este biosimilar. Los médicos han enviado información a este registro de 818 pacientes tratados con BEVZ92 entre 2016 y 2018 por cáncer colorrectal metastásico, cáncer de ovario epitelial, cáncer de cuello uterino recurrente, metastásico o persistente, cáncer de mama metastásico, cáncer de pulmón de células no pequeñas avanzado, glioblastoma, cáncer avanzado o carcinoma metastásico de células renales e indicaciones clínicas de cáncer fuera de etiqueta [ 101]. De ellos, se incluyeron para el análisis 416 pacientes que tenían al menos un punto de seguimiento, con 44 informes presentados que involucraban 51 RAM (23 graves). La comparación de la lista de reacciones adversas en pacientes con cáncer para BEVZ92 con las del bevacizumab de referencia en estudios de vigilancia posteriores a la comercialización muestra similitud con el anticuerpo de referencia, pero el número relativamente bajo de informes enfatiza la necesidad de continuar con este programa de farmacovigilancia para establecer mejor el perfil de seguridad de BEVZ92 en pacientes con cáncer [ 101 ].
2.5.2 Mvasi® (Amgen)
ABP 215 (Mvasi®) es un biosimilar de bevacizumab desarrollado por Amgen en los EE. UU. Fue el primer biosimilar de este anticuerpo monoclonal original aprobado por la FDA en 2017 y por la EMA en 2018 [ 69 ]. ABP 215 está indicado para el tratamiento de pacientes con carcinoma metastásico de colon o recto, cáncer de mama metastásico, cáncer de pulmón de células no pequeñas metastásico o recurrente, cáncer de células renales avanzado y/o metastásico y cáncer epitelial de ovario, trompa de Falopio, peritoneal primario o cáncer de cuello uterino ABP 215 es comercializado por Amgen en Argentina, Brasil y Colombia con la marca Mvasi®.
Las pruebas analíticas para evaluar la similitud entre ABP 215 y el bevacizumab original demostraron que ambos productos tienen la misma secuencia peptídica y que el perfil de glicosilación era similar. Las actividades biológicas y funcionales de ABP 215 y bevacizumab de referencia mostraron una unión e inhibición similares de la señalización de VEGFR-2 entre los grupos. Se evaluó la similitud de más de 20 lotes de bevacizumab original y 13 lotes de ABP 215 y se demostró que los atributos estructurales y de pureza y las propiedades biológicas son muy similares entre ellos [ 102 ].
Para evaluar la equivalencia de farmacocinética, seguridad, tolerabilidad e inmunogenicidad del biosimilar ABP 215 y el bevacizumab de referencia, se realizó un estudio clínico de fase I aleatorizado, simple ciego, de dosis única. En este ensayo, la concentración sérica máxima observada y el AUC fueron similares entre ABP 215 y el bevacizumab de referencia. Además, los perfiles de seguridad no mostraron diferencias, sin muertes ni eventos adversos que condujeran a la interrupción del estudio, y ningún sujeto dio positivo para la unión de anticuerpos antidrogas [ 61 ].
La equivalencia clínica en términos de seguridad, inmunogenicidad y eficacia entre ABP 215 y el bevacizumab original se evaluó en un ensayo clínico de fase III en pacientes con cáncer de pulmón no microcítico no escamoso avanzado. La frecuencia, el tipo y la gravedad de los eventos adversos fueron comparables entre ABP 215 y bevacizumab de referencia, y ningún paciente dio positivo en anticuerpos neutralizantes antidrogas. Además, la eficacia clínica de ABP 215 y el bevacizumab de referencia fue similar, con una respuesta global del paciente del 39,0 y el 41,7 %, respectivamente. Los datos de este ensayo clínico respaldan una equivalencia clínica ABP 215 y bevacizumab original [ 103 ].
3. Conclusión
En la última década, los avances en las vías regulatorias para registrar medicamentos biológicos con estándares de muy alta calidad permitieron la aprobación de la primera generación de anticuerpos monoclonales biosimilares en América Latina que mostró evidencia robusta de seguridad y eficacia. Este proceso se produjo en paralelo con la expiración de las patentes de los primeros anticuerpos monoclonales terapéuticos. Para fines de 2021, se espera comercializar en la región anticuerpos biosimilares de rituximab, trastuzumab, infliximab, adalimumab y bevacizumab con 25 marcas diferentes. Esta tendencia es más fuerte en países como Brasil y Argentina, que tienen más de diez anticuerpos monoclonales biosimilares diferentes aprobados y, como es el caso de trastuzumab, tres biosimilares diferentes aprobados que compiten con Herceptin®, el anticuerpo de referencia.
Recibido: 10 de febrero de 2021 Revisado: 14 de octubre de 2021 Publicado: 7 de diciembre de 2021
DOI: 10.5772/intechopen.101227